"The transition from relying on public data to independently establishing high-quality datasets is the key to improving model accuracy," said Li Feiran.
She has been long committed to the research of metabolic network models, involving computational biology, systems biology, machine learning, chemistry, drug metabolism, and other fields. She has obtained a Ph.D. in Bioscience and Bioengineering from Chalmers University of Technology in Sweden, under the guidance of the world-renowned bioengineering expert Professor Jens Nielsen.
Currently, Li Feiran is one of the scientists collaborating with Jushu Bio to build a "precision" enzyme engineering model, serving as an assistant professor and a special researcher at the Shenzhen International Graduate School of Tsinghua University.
"The core of AI-assisted 'precision' enzyme engineering design lies in high-quality datasets. Based on high-throughput activity testing of typical industrial enzymes, proprietary datasets are obtained, combined with deep learning models to achieve standardization and precision of enzyme engineering design. Jushu Bio is moving in this direction," said Professor Zhang Chong from Tsinghua University, the scientific founder of Jushu Bio.
Advertisement
The first "large model" for enzyme activity predictionEnzymes are not only involved in the biomanufacturing process, but they are also an important product of biomanufacturing. In 2023, the global market size of industrial enzymes alone reached 7.4 billion US dollars.
Molecular design and modification of enzyme proteins are key to creating high-performance industrial enzymes, reducing production costs, and enhancing industrial competitiveness. Enzyme engineering mainly includes strategies such as rational design, directed evolution, semi-rational design, and artificial intelligence-assisted design.
Among them, under the drive of data, AI technology can learn about the characteristics of protein composition and evolution, and it can solve many types of enzyme engineering problems. For example, predicting mutations with beneficial effects, optimizing protein stability, and improving catalytic activity.
However, AI currently faces many challenges in protein design. In the aspect of enzyme modification design, AI finds it difficult to accurately predict the structural changes caused by minor perturbations [1-3].
In the de novo design of enzymes, the challenges faced by AI are even more complex.The success rate of generating world-leading models with specific structures is 15-50%, while the success rate for generating world-leading models with specific functions is about 0.01%-60% (for soluble simple proteins), and the success rate for generating highly active world-leading models is far less than 10% [4-6].
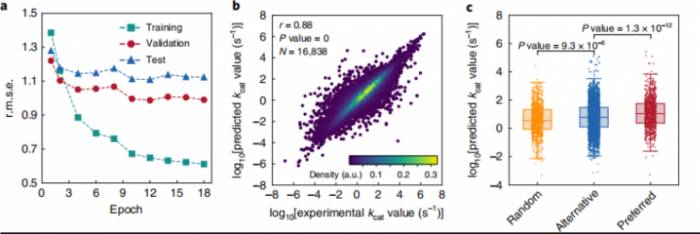
Li Feiran and his collaborators have developed for the first time a deep learning model called DLKcat and an enzyme parameter database called GotEnzymes, which can achieve large-scale enzyme activity characterization.
kcat (catalytic turnover number) is an important kinetic constant for understanding the catalytic properties of enzymes. The DLKcat deep learning model can successfully predict the enzyme activity parameter kcat, and by inputting substrate SMILES information and the protein sequence of the enzyme, one can obtain intuitive specific enzyme kinetic parameters.
"Without data-driven or AI models, this is very difficult to achieve," said Li Feiran.
It is understood that the enzyme parameter database GotEnzymes, also created by Li Feiran, has predicted enzyme activity parameters for most known enzymes to date. Users can obtain high-activity candidate enzymes with specific functions through simple queries.The first version of GotEnzymes covers the vast majority of enzyme classes, including: over 5.8 million types of enzymes and 25.79 million predicted turnover numbers for enzyme-compound pairs.
Moreover, each pair is labeled with an EC number (Enzyme Commission number, also known as the enzyme nomenclature), covering 8,099 organisms, including 747 eukaryotes, 6,963 bacteria, and 389 archaea.
It can be visually observed that eukaryotes generally have a higher turnover number per organism. The median turnover number for the entire dataset is 5s^-1, and most values (75%) fall between 1 and 100s^-1, which is consistent with studies based on experimental data.
When grouped by organism, it is found that the median turnover numbers for eukaryotes, bacteria, and archaea are close, with the median for eukaryotes being the lowest. When grouped by EC number, it is found that isomerases (EC 5.X.X.X) have the highest median, while ligases (EC 6.X.X.X) have the lowest, which is consistent with previous findings.
The performance of GotEnzymes will be continuously improved through an iterative mechanism. By using code version control on GitHub and a reproducible prediction pipeline, data can be regenerated at future time points to extend predictions for other enzymes and other parameters.AI tools, through continuous iteration, can achieve better predictions with more and better training data. Therefore, it is expected that GotEnzymes will improve its performance as the training data increases.
Moreover, with the deployment of improved algorithms in the future, the prediction module of GotEnzymes for predicting different types of parameters can be updated independently, quickly releasing updated versions of the database.
Ultimately, the relevant papers were published with the titles "Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction" in Nature Catalysis[7], and "GotEnzymes: an extensive database of enzyme parameter predictions" in Nucleic Acids Research[8].
The first paper was featured and recommended by Nature Catalysis in the News & Views column. In addition, due to the broad application scenarios and importance of machine learning in the field of catalysis, this paper was selected as one of the 12 focus papers in the "Machine Learning in Catalysis" column of Nature Catalysis[9].Professor Costas Maranas from Pennsylvania State University, an expert in metabolic modeling, praised DLKcat as "capable of predicting the activity of any metabolic enzyme" [10].
The second paper was evaluated by George Church, a member of the National Academy of Sciences and an expert in synthetic biology, as "machine learning compensates for the lack of parameters in mechanism models, aiding in the design of the next generation of cell factories" [11].
Professor Xu Jianhe from East China University of Science and Technology, an expert in the field of enzyme engineering, stated that this artificial intelligence research result is of milestone significance. Its core content successfully constructs a method based on deep learning (DLKcat), which can predict kcat from the sequence and substrate structure of enzymes. It can be used to reconstruct enzyme capacity-constrained genome-scale metabolic models, especially suitable for characterizing the metabolic processes of yeast and fungal cells.
Screen 100 enzymes in 1 second.It is worth noting that the aforementioned kcat prediction method will significantly enhance the efficiency of enzyme engineering. For a specific class of industrial enzymes, if more standardized experimental data are used for iterative training in the future, it is believed that the prediction accuracy and applicability of DLKcat will be increasingly perfected.
According to the introduction, the published DLKcat is the first version, and the accuracy error of the enzyme activity prediction model is within one order of magnitude, which already has guiding significance for the field of enzyme engineering and enzyme design.
Through continuous updating and iteration, the GotEnzymes database based on DLKcat has now evolved to the second version. It can not only predict enzyme activity but also achieve predictions of various indicators such as enzyme affinity, optimal temperature, with significantly improved accuracy.
In addition, it can also perform some simulated structural modifications of enzymes (such as adding proline, shortening, adding conserved sequences, etc.) semi-generative enzyme modifications to optimize enzyme characteristics (such as temperature stability).
"At present, the level of our second version of DLKcat and GotEnzymes is at the leading level in the industry," said Li Feiran.She stated that the database significantly reduces the technical difficulty for researchers who are not proficient in programming. They can directly find the target enzymes on the website without spending time and effort learning how to build deep learning models from scratch.
Following this technological breakthrough, there has been a surge of interest in the prediction of enzyme activity and enzyme parameters within the field. Subsequently, more than ten research groups have also reported related studies on the prediction of enzyme parameters.
In addition, several research groups have utilized the GotEnzymes database. For example, the team led by Academician Yuan Yingjin from Tianjin University used GotEnzymes to predict enzyme activity [12].
Li Feiran said: "This model has low computational requirements for the end device, and can even be used directly on a laptop. Moreover, it can screen at least 100 enzymes in one second."
This technology provides an important tool for the field of synthetic biology, not only reducing the randomness of selection but also offering more rational design of enzyme selectivity.From an application perspective, in the short term, people can directly use this model to predict for any enzyme and any substrate. Moreover, it is expected to achieve customized enzyme components with high activity, specific structure, or resistance to high temperature and strong acid environments.
Further, by continuously introducing the relevant parameters of this technology into the modeling of digital life in systems biology, it is conducive to better simulating cell phenotypes for the design of cell factories and the analysis of the mechanisms of evolutionary diseases.
It is worth noting that in the application promotion of enzyme design or enzyme characterization tools, the key lies in continuous iteration to continuously improve the performance of the tools.
In fact, the models trained based on public data currently face the same challenges in prediction accuracy and there is no essential difference. In the future, training more accurate enzyme design and characterization tools with more proprietary data will have stronger industry competitiveness and higher commercial value.
It is understood that at present, the startup company Jushu Biology has established in-depth cooperation with Li Feiran to jointly develop a "precise" large model of enzyme engineering.When discussing this research, Zhang Chong stated that based on the high-throughput enzyme engineering modification and activity detection technology platform, high-quality specialized enzyme activity feature datasets will be constructed at low cost and quickly, providing precise and annotated data for deep learning models. This will enable deep learning models to achieve higher accuracy in the field of enzyme design, realizing the "data-model" flywheel effect in the enzyme design field.
How to produce high-quality specialized enzyme data?
Zhang Chong said that for new enzyme sequences output by AI big data models, it is necessary to systematically characterize the actual performance of the enzymes, screen for "high-quality enzyme sequences," verify the accuracy of AI models, and further calibrate and improve AI models through testing and optimization data. To achieve the above goals, two main steps need to be completed: "new enzyme expression" and "new enzyme testing."
Traditional methods of sequence synthesis, chassis cell transformation, cultivation, protein purification, and enzyme activity detection have always faced the challenges of low manual efficiency and high experimental costs, and cannot high-throughput verify the performance and process parameters of new enzyme sequences produced by AI data models.The autoHIPPS system developed by the Jushu Biological Team is based on a droplet microfluidics + automated robotic arm equipment "high-throughput molecular cloning - high-throughput single-cell culture - engineering enzyme preparation and purification - rapid enzyme activity detection" automated experimental process, which can meet the "new enzyme sequence" high-throughput, low-cost preparation and enzyme characteristic activity evaluation and screening of the entire process.
Each step of this process can achieve a test volume of 10^3 to 10^6 samples/day, obtaining high-throughput "high-quality enzyme sequence" corresponding to "characteristic enzyme activity parameters", which are used to optimize the AI big data model of this class of enzymes, and quickly improve the accuracy of high-quality sequence prediction.
Li Feiran said that the autoHIPPS system provides a large amount of high-quality data for AI enzyme parameter prediction through a high-throughput, low-cost, fully automated experimental process, which can significantly improve the prediction accuracy and efficiency of the AI model.
It is understood that the autoHIPPS system is based on the "cell culture - protein expression - purification preparation - automated detection" process of the automated workstation module, which can achieve the "rapid preparation - purification - detection" of the new enzyme sequence entity protein output by the AI model, and reduce the cost of culture and enzyme purification by more than 90%, and shorten the enzyme performance test pretreatment time from 1 hour to 10 seconds.
Based on the standard enzyme activity detection method, the entity protein of the new enzyme sequence is subjected to high-throughput performance testing and evaluation (catalytic activity, substrate specificity, thermal stability, etc.). While obtaining the best enzyme sequence, a test data set of 10^4 to 10^6 samples/day is achieved, providing high-accuracy quality data for further optimization of the AI big model.Zhang Chong stated that through the autoHIPPS ultra-high-throughput droplet microfluidics + robotic arm automation equipment platform, the "high-throughput solid protein preparation, testing, and evaluation" of new enzymes can be realized. The high-quality proprietary enzyme database formed thereby can enrich the assessment and prediction dimensions of AI new enzyme models, accelerating model iteration and optimization.
Goal: Cover the entire chain from enzyme design to providing enzyme products.
As the most comprehensive and widely used enzyme information resource, the BRENDA enzyme database has been collecting enzyme parameters for decades. As of January 2022, there are 83,662 turnover numbers in BRENDA, far less than the number calculated and predicted in GotEnzymes.
Due to the large amount of data, Li Feiran said that GotEnzymes will be able to provide a speed-up for biological research, including experimental and computational fields.On one hand, GotEnzymes is poised to provide the best enzymes based on predictive parameters, guiding the selection and design of enzymes, thereby reducing the time of the "design-build-test-learn" cycle in synthetic biology and metabolic engineering.
On the other hand, GotEnzymes facilitates computational analysis across organisms through its Application Programming Interface (API). For instance, evolutionary analysis and metabolic modeling that relies on large-scale enzyme parameters, such as kinetic models and proteome constraint models.
Next, the team will expand GotEnzymes by integrating more types of enzyme parameters, leveraging available AI-based predictions, such as the optimal temperature for enzymes and Michaelis constants, to meet more of the users' needs.
In addition, they will also implement annotations from other databases like MetaCyc and annotation tools based on deep learning, to expand the coverage of enzyme-compound pairs initially generated based solely on the KEGG database.
At the same time, researchers also intend to overlay enzyme parameters as a new layer on the metabolic pathway maps on the Metabolic Atlas platform, which is expected to enable interactive comparisons and facilitate the development of advanced models.After obtaining high-performance enzyme sequences, efficient expression can be achieved through data-mechanism hybrid-driven chassis cell design technology, thereby realizing the full chain transition from design to production.
"In the future, after users propose specific enzyme design requirements, we will be able to directly provide a full chain of services from design and modification to expression, and then to enzyme products," said Li Feiran.
